


Keywords
chemical reaction
chemoinformatics
QSAR
QSPR
QSRR
reaction conditions
reaction informatics
reaction rate
reaction yield
Abstract
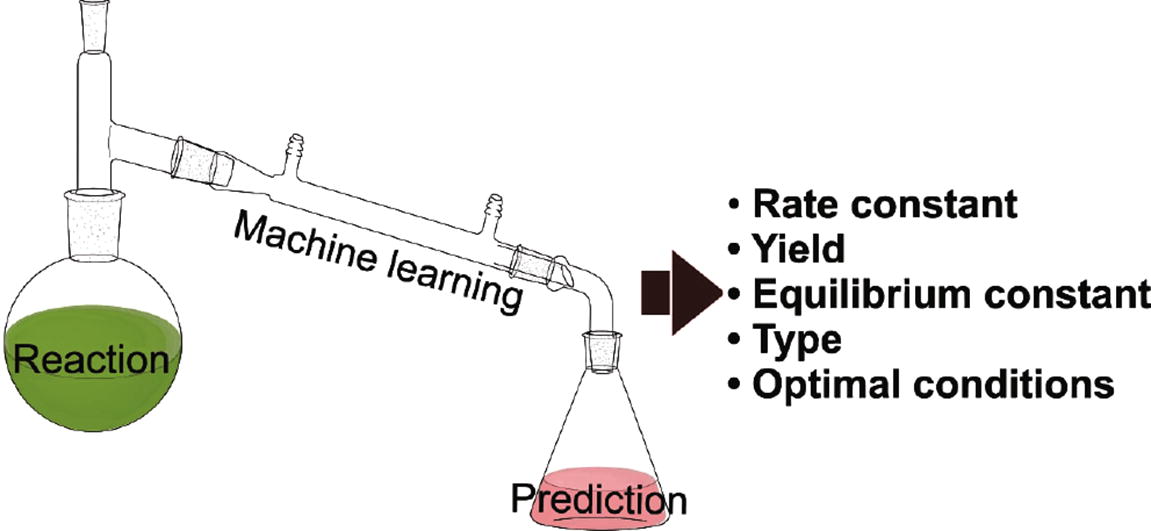
The synthesis of the desired chemical compound is the main task of synthetic organic chemistry. The predictions of reaction conditions and some important quantitative characteristics of chemical reactions as yield and reaction rate can substantially help in the development of optimal synthetic routes and assessment of synthesis cost. Theoretical assessment of these parameters can be performed with the help of modern machine-learning approaches, which use available experimental data to develop predictive models called quantitative or qualitative structure–reactivity relationship (QSRR) modelling. In the article, we review the state-of-the-art in the QSRR area and give our opinion on emerging trends in this field.
References
1.
Corey E.J.
Chemical Society Reviews,
1988
2.

Baskin I.I., Madzhidov T.I., Antipin I.S., Varnek A.A.
Russian Chemical Reviews,
2017
3.
Schwaller P., Petraglia R., Zullo V., Nair V.H., Haeuselmann R.A., Pisoni R., Bekas C., Iuliano A., Laino T.
Chemical Science,
2020
4.

Segler M.H., Preuss M., Waller M.P.
Nature,
2018
5.

Coley C.W., Green W.H., Jensen K.F.
Accounts of Chemical Research,
2018
6.

Engkvist O., Norrby P., Selmi N., Lam Y., Peng Z., Sherer E.C., Amberg W., Erhard T., Smyth L.A.
Drug Discovery Today,
2018
7.

Szymkuć S., Gajewska E.P., Klucznik T., Molga K., Dittwald P., Startek M., Bajczyk M., Grzybowski B.A.
Angewandte Chemie - International Edition,
2016
8.
Hammett L.P.
Journal of the American Chemical Society,
1937
9.
10.1016/j.mencom.2021.11.003_b0045
Hammond
Pure Appl. Chem.,
1919
10.
10.1016/j.mencom.2021.11.003_b0050
Anslyn
Modern Physical Organic Chemistry,
2006
11.
Taft R.W.
Journal of the American Chemical Society,
1952
12.
10.1016/j.mencom.2021.11.003_b0060
Palm
Khimiya,
1977
13.
14.
Hansch C., Fujita T.
Journal of the American Chemical Society,
1964
15.
Rekker R.F.
Quantitative Structure-Activity Relationships,
1992
16.
Ignatz-Hoover F., Petrukhin R., Karelson M., Katritzky A.R.
Journal of Chemical Information and Computer Sciences,
2001
17.
Chaudry U.A., Popelier P.L.
Journal of Physical Chemistry A,
2003
18.
Zhang H., Qu X., Ando H.
Journal of Molecular Structure THEOCHEM,
2005
19.
Katritzky A.R., Perumal S., Petrukhin R.
Journal of Organic Chemistry,
2001
20.
G. R. Famini and L. Y. Wilson, in Reviews in Computational Chemistry, eds. K. B. Lipkowitz and D. B. Boyd, John Wiley & Sons, 2003, vol. 18, pp. 211–255.
21.
Hansch C., Leo A., Taft R.W.
Chemical Reviews,
1991
22.
10.1016/j.mencom.2021.11.003_b0110
2003
23.
Halberstam N.M., Baskin I.I., Palyulin V.A., Zefirov N.S.
Mendeleev Communications,
2002
24.
D. M. Lowe, PhD Thesis, 2012, doi: https://doi.org/10.17863/ CAM.16293.
25.

Baskin I.I., Winkler D., Tetko I.V.
Expert Opinion on Drug Discovery,
2016
26.
Schwaller P., Gaudin T., Lányi D., Bekas C., Laino T.
Chemical Science,
2018
27.
Schwaller P., Laino T., Gaudin T., Bolgar P., Hunter C.A., Bekas C., Lee A.A.
ACS Central Science,
2019
28.
Lin A.I., Madzhidov T.I., Klimchuk O., Nugmanov R.I., Antipin I.S., Varnek A.
Journal of Chemical Information and Modeling,
2016
29.
10.1016/j.mencom.2021.11.003_b0145
Gao
Sci.,
2018
30.
Bort W., Baskin I.I., Gimadiev T., Mukanov A., Nugmanov R., Sidorov P., Marcou G., Horvath D., Klimchuk O., Madzhidov T., Varnek A.
Scientific Reports,
2021
31.
Muratov E.N., Bajorath J., Sheridan R.P., Tetko I.V., Filimonov D., Poroikov V., Oprea T.I., Baskin I.I., Varnek A., Roitberg A., Isayev O., Curtalolo S., Fourches D., Cohen Y., Aspuru-Guzik A., et. al.
Chemical Society Reviews,
2020
32.
10.1016/j.mencom.2021.11.003_b0160
Extance
Chem. World,
2020
33.

Coley C.W., Thomas D.A., Lummiss J.A., Jaworski J.N., Breen C.P., Schultz V., Hart T., Fishman J.S., Rogers L., Gao H., Hicklin R.W., Plehiers P.P., Byington J., Piotti J.S., Green W.H., et. al.
Science,
2019
34.
Chemoinformatics: A Textbook, eds. J. Gasteiger and T. Engel, Wiley- VCH, Weinheim, 2003.
35.
Weininger D.
Journal of Chemical Information and Modeling,
1988
36.
Dalby A., Nourse J.G., Hounshell W.D., Gushurst A.K., Grier D.L., Leland B.A., Laufer J.
Journal of Chemical Information and Computer Sciences,
1992
37.
38.
Daylight Theory Manual, version 4.1, Daylight Chemical Information Systems, Laguna Niguel, CA, 2011, https://www.daylight.com/ dayhtml/doc/theory/index.
39.
10.1016/j.mencom.2021.11.003_b0195
Dugundji
Top. Curr. Chem.,
1973
40.
41.
Gasteiger J., Jochum C.
42.
Gasteiger J., Hutchings M.G., Christoph B., Gann L., Hiller C., Löw P., Marsili M., Saller H., Yuki K.
Topics in Current Chemistry,
1987
43.
10.1016/j.mencom.2021.11.003_b0215
Chen
Wiley Interdiscip. Rev.: Comput. Mol. Sci.,
2013
44.
Varnek A., Fourches D., Hoonakker F., Solov’ev V.P.
Journal of Computer-Aided Molecular Design,
2005
45.
Nugmanov R.I., Mukhametgaleev R.N., Akhmetshin T., Gimadiev T.R., Afonina V.A., Madzhidov T.I., Varnek A.
Journal of Chemical Information and Modeling,
2019
46.
A. Wagner, F. Hoonakker and A. Varnek, US Patent 2009/0024575 A1, 2009.
47.
de Luca A., Horvath D., Marcou G., Solov’ev V., Varnek A.
Journal of Chemical Information and Modeling,
2012
48.
Delannée V., Nicklaus M.C.
Journal of Cheminformatics,
2020
49.
Glavatskikh M., Madzhidov T., Baskin I.I., Horvath D., Nugmanov R., Gimadiev T., Marcou G., Varnek A.
Molecular Informatics,
2018
50.
Gimadiev T., Madzhidov T., Tetko I., Nugmanov R., Casciuc I., Klimchuk O., Bodrov A., Polishchuk P., Antipin I., Varnek A.
Molecular Informatics,
2018
51.

Nugmanov R.I., Madzhidov T.I., Khaliullina G.R., Baskin I.I., Antipin I.S., Varnek A.A.
Journal of Structural Chemistry,
2014
52.
Madzhidov T.I., Bodrov A.V., Gimadiev T.R., Nugmanov R.I., Antipin I.S., Varnek A.A.
Journal of Structural Chemistry,
2015
53.
Kravtsov A.A., Karpov P.V., Baskin I.I., Palyulin V.A., Zefirov N.S.
Doklady Chemistry,
2011
54.
Kravtsov A.A., Karpov P.V., Baskin I.I., Palyulin V.A., Zefirov N.S.
Doklady Chemistry,
2011
55.
Sandfort F., Strieth-Kalthoff F., Kühnemund M., Beecks C., Glorius F.
Chem,
2020
56.
Marcou G., Aires de Sousa J., Latino D.A., de Luca A., Horvath D., Rietsch V., Varnek A.
Journal of Chemical Information and Modeling,
2015
57.
Polishchuk P., Madzhidov T., Gimadiev T., Bodrov A., Nugmanov R., Varnek A.
Journal of Computer-Aided Molecular Design,
2017
58.
Schneider N., Lowe D.M., Sayle R.A., Landrum G.A.
Journal of Chemical Information and Modeling,
2015
59.
10.1016/j.mencom.2021.11.003_b0295
Hu
PLoS One,
2012
60.
Zhang Q., Aires-de-Sousa J.
Journal of Chemical Information and Modeling,
2005
61.

Latino D.A., Zhang Q., Aires-de-Sousa J.
Bioinformatics,
2008
62.
Faulon J., Misra M., Martin S., Sale K., Sapra R.
Bioinformatics,
2007
63.
Ridder L., Wagener M.
ChemMedChem,
2008
64.
Oprisiu I., Varlamova E., Muratov E., Artemenko A., Marcou G., Polishchuk P., Kuz'min V., Varnek A.
Molecular Informatics,
2012
65.
Varnek A., Fourches D., Horvath D., Klimchuk O., Gaudin C., Vayer P., Solov'ev V., Hoonakker F., Tetko I., Marcou G.
Current Computer-Aided Drug Design,
2008
66.
67.
Horvath D., Marcou G., Varnek A., Kayastha S., de la Vega de León A., Bajorath J.
Journal of Chemical Information and Modeling,
2016
68.
Glavatskikh M., Madzhidov T., Horvath D., Nugmanov R., Gimadiev T., Malakhova D., Marcou G., Varnek A.
Molecular Informatics,
2018
69.
Madzhidov T.I., Gimadiev T.R., Malakhova D.A., Nugmanov R.I., Baskin I.I., Antipin I.S., Varnek A.A.
Journal of Structural Chemistry,
2017
70.
Gimadiev T.R., Madzhidov T.I., Nugmanov R.I., Baskin I.I., Antipin I.S., Varnek A.
Journal of Computer-Aided Molecular Design,
2018
71.
Catalán J., López V., Pérez P., Martin-Villamil R., Rodríguez J.
European Journal of Organic Chemistry,
1995
72.
Catalán J., Díaz C.
European Journal of Organic Chemistry,
1997
73.
Catalán J., Díaz C.
European Journal of Organic Chemistry,
1999
74.
Catalán J., Díaz C., López V., Pérez P., De Paz J.G., Rodríguez J.G.
European Journal of Organic Chemistry,
1996
75.
Kamlet M.J., Taft R.W.
Journal of the American Chemical Society,
1976
76.
Taft R.W., Kamlet M.J.
Journal of the American Chemical Society,
1976
77.
Kamlet M.J., Abboud J.L., Taft R.W.
Journal of the American Chemical Society,
1977
78.
10.1016/j.mencom.2021.11.003_b0390
Marcus
The Properties of Solvents,
1998
79.
Skoraczyński G., Dittwald P., Miasojedow B., Szymkuć S., Gajewska E.P., Grzybowski B.A., Gambin A.
Scientific Reports,
2017
80.
10.1016/j.mencom.2021.11.003_b0400
Rakhimbekova
SARQSAR Environ. Res.,
2021
81.
React. – CASREACT, 2021, http://www.cas.org/support/documentation/ reactions.
82.
Reaxys, 2021, www. reaxys.com.
83.
Goodman J.
Journal of Chemical Information and Modeling,
2009
84.
SPRESI, 2019, http://www.spresi.com/.
85.
SciVal, 2021, https://www.scival.com/.
86.
Gimadiev T.R., Lin A., Afonina V.A., Batyrshin D., Nugmanov R.I., Akhmetshin T., Sidorov P., Duybankova N., Verhoeven J., Wegner J., Ceulemans H., Gedich A., Madzhidov T.I., Varnek A.
Molecular Informatics,
2021
87.
Pistachio, 2021, https://www.nextmovesoftware.com/pistachio.html.
88.
W. Jin, C.W. Coley, R. Barzilay and T. Jaakkola, arXiv: 1709.04555, 2017.
89.
Schneider N., Stiefl N., Landrum G.A.
Journal of Chemical Information and Modeling,
2016
90.
10.1016/j.mencom.2021.11.003_b0450
Nguyen
Advances in Information Retrieval,
2020
91.
Ahneman D.T., Estrada J.G., Lin S., Dreher S.D., Doyle A.G.
Science,
2018
92.
Tables of Rate and Equilibrium Constants of Heterolytic Organic Reactions, ed. V. I. Palm, VINITI, 1978.
93.
ChemInform Reaction Library, 2021, http://www.cheminform.com/ reaction.
94.
W. Jin and C. W. Coley, Rexgen, 2021, https://github.com/wengong-jin/nips17-rexgen.
95.
Hammett L.P.
Chemical Reviews,
1935
96.
Hammett L.P.
Transactions of the Faraday Society,
1938
97.
McDuffie H.F., Dougherty G.
Journal of the American Chemical Society,
1942
98.
Zhang Q., Qu X., Wang H., Xu F., Shi X., Wang W.
Environmental Science & Technology,
2009
99.
Bräuer M., Pérez-Lustres J.L., Weston J., Anders E.
Inorganic Chemistry,
2002
100.
10.1016/j.mencom.2021.11.003_b0500
Advances in Linear Free Energy Relationships,
1972
101.
Zhokhova N.I., Baskin I.I., Palyulin V.A., Zefirov A.N., Zefirov N.S.
Doklady Chemistry,
2007
102.

HOONAKKER F., LACHICHE N., VARNEK A., WAGNER A.
International Journal on Artificial Intelligence Tools,
2011
103.

Rakhimbekova A., Madzhidov T.I., Nugmanov R.I., Gimadiev T.R., Baskin I.I., Varnek A.
International Journal of Molecular Sciences,
2020
104.
Tetko I.V., Sushko I., Pandey A.K., Zhu H., Tropsha A., Papa E., Öberg T., Todeschini R., Fourches D., Varnek A.
Journal of Chemical Information and Modeling,
2008
105.
Bergman R.G., Danheiser R.L.
Angewandte Chemie - International Edition,
2016
106.
107.

Schwaller P., Vaucher A.C., Laino T., Reymond J.
Machine Learning: Science and Technology,
2021
108.
Fu Z., Li X., Wang Z., Li Z., Liu X., Wu X., Zhao J., Ding X., Wan X., Zhong F., Wang D., Luo X., Chen K., Liu H., Wang J., et. al.
Organic Chemistry Frontiers,
2020
109.
Granda J.M., Donina L., Dragone V., Long D., Cronin L.
Nature,
2018
110.
Nielsen M.K., Ahneman D.T., Riera O., Doyle A.G.
Journal of the American Chemical Society,
2018
111.
Chuang K.V., Keiser M.J.
Science,
2018
112.
Perera D., Tucker J.W., Brahmbhatt S., Helal C.J., Chong A., Farrell W., Richardson P., Sach N.W.
Science,
2018
113.
Reizman B.J., Wang Y., Buchwald S.L., Jensen K.F.
Reaction Chemistry and Engineering,
2016
114.
A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, L. Kaiser and I. Polosukhin, arXiv: 1706.03762, 2017.
115.
Schwaller P., Probst D., Vaucher A.C., Nair V.H., Kreutter D., Laino T., Reymond J.
Nature Machine Intelligence,
2021
116.
Mansouri K., Cariello N.F., Korotcov A., Tkachenko V., Grulke C.M., Sprankle C.S., Allen D., Casey W.M., Kleinstreuer N.C., Williams A.J.
Journal of Cheminformatics,
2019
117.
10.1016/j.mencom.2021.11.003_b0590
Lee
J. Chem. Inf. Model.,
2013
118.
Luan F., Ma W., Zhang H., Zhang X., Liu M., Hu Z., Fan B.
Pharmaceutical Research,
2005
119.
Jensen J.H., Swain C.J., Olsen L.
Journal of Physical Chemistry A,
2017
120.
Eckert F., Klamt A.
Journal of Computational Chemistry,
2005
121.
Liao C., Nicklaus M.C.
Journal of Chemical Information and Modeling,
2009
122.
10.1016/j.mencom.2021.11.003_b0615
Elguero
The Tautomerism of Heterocycles,
1976
123.
Klamt A., Diedenhofen M.
Journal of Computer-Aided Molecular Design,
2010
124.
Soteras I., Orozco M., Luque F.J.
Journal of Computer-Aided Molecular Design,
2010
125.
Greenwood J.R., Calkins D., Sullivan A.P., Shelley J.C.
Journal of Computer-Aided Molecular Design,
2010
126.
Alkorta I., Elguero J.
Journal of Physical Organic Chemistry,
2005
127.
J. Szegezdi and F. Csizmadia, in Fall ACS National Meeting, Boston, August 19–23, 2007.
128.
Milletti F., Storchi L., Sforna G., Cross S., Cruciani G.
Journal of Chemical Information and Modeling,
2009
129.
Zankov D.V., Madzhidov T.I., Rakhimbekova A., Gimadiev T.R., Nugmanov R.I., Kazymova M.A., Baskin I.I., Varnek A.
Journal of Chemical Information and Modeling,
2019
130.
Kozlowski M.C., Dixon S.L., Panda M., Lauri G.
Journal of the American Chemical Society,
2003
131.
Zahrt A.F., Athavale S.V., Denmark S.E.
Chemical Reviews,
2019
132.
Woolfrey J.R., Avery M.A., Doweyko A.M.
Journal of Computer-Aided Molecular Design,
1998
133.
3D QSAR in Drug Design, ed. H. Kubinyi, Springer, Netherlands, 1994.
134.
Cramer R.D., Patterson D.E., Bunce J.D.
Journal of the American Chemical Society,
1988
135.
Lipkowitz K.B., Pradhan M.
Journal of Organic Chemistry,
2003
136.
Pastor M., Cruciani G., McLay I., Pickett S., Clementi S.
Journal of Medicinal Chemistry,
2000
137.
Sciabola S., Alex A., Higginson P.D., Mitchell J.C., Snowden M.J., Morao I.
Journal of Organic Chemistry,
2005
138.
Braiuca P., Lorena K., Ferrario V., Ebert C., Gardossi L.
Advanced Synthesis and Catalysis,
2009
139.
Harper K.C., Bess E.N., Sigman M.S.
Nature Chemistry,
2012
140.
Miller J., Sigman M.
Angewandte Chemie - International Edition,
2008
141.
Oslob J.D., Åkermark B., Helquist P., Norrby P.
Organometallics,
1997
142.
Metsänen T.T., Lexa K.W., Santiago C.B., Chung C.K., Xu Y., Liu Z., Humphrey G.R., Ruck R.T., Sherer E.C., Sigman M.S.
Chemical Science,
2018
143.
Park Y., Niemeyer Z.L., Yu J., Sigman M.S.
Organometallics,
2017
144.
Melville J.L., Lovelock K.R., Wilson C., Allbutt B., Burke E.K., Lygo B., Hirst J.D.
Journal of Chemical Information and Modeling,
2005
145.
Zahrt A.F., Henle J.J., Rose B.T., Wang Y., Darrow W.T., Denmark S.E.
Science,
2019
146.
Henle J.J., Zahrt A.F., Rose B.T., Darrow W.T., Wang Y., Denmark S.E.
Journal of the American Chemical Society,
2020
147.
148.
10.1016/j.mencom.2021.11.003_b0750
Xu
Synlett,
2020
149.
10.1016/j.mencom.2021.11.003_b0755
Zankov
Synlett,
2021
150.
Kutlushina A., Khakimova A., Madzhidov T., Polishchuk P.
Molecules,
2018
151.
D. V. Zankov, M. Matveieva, A. Nikonenko, R. Nugmanov, A. Varnek, P. Polishchuk and T. Madzhidov, ChemRxiv Prepr. 13456277, 2020, 1.
152.
153.
10.1016/j.mencom.2021.11.003_b0775
Balaban
Rev. Roum. Chim.,
1967
154.
155.
156.
A formalism for the classification and design of organic reactions. I. The class of (− +)n reactions
Arens J.F.
Recueil des Travaux Chimiques des Pays-Bas,
1979
157.
Tratch S.S., Zefirov N.S.
Journal of Chemical Information and Computer Sciences,
1998
158.
10.1016/j.mencom.2021.11.003_b0800
Zefirov
MATCH,
1977
159.
Fujita S.
Journal of Chemical Information and Computer Sciences,
1986
160.
Fujita S.
Journal of Chemical Information and Computer Sciences,
1987
161.
Kraut H., Eiblmaier J., Grethe G., Löw P., Matuszczyk H., Saller H.
Journal of Chemical Information and Modeling,
2013
162.
Roughley S.D., Jordan A.M.
Journal of Medicinal Chemistry,
2011
163.
Carey J.S., Laffan D., Thomson C., Williams M.T.
Organic and Biomolecular Chemistry,
2006
164.
10.1016/j.mencom.2021.11.003_b0830
NextMove Software,
2020
165.
Schneider N., Lowe D.M., Sayle R.A., Tarselli M.A., Landrum G.A.
Journal of Medicinal Chemistry,
2016
166.
Christ C.D., Zentgraf M., Kriegl J.M.
Journal of Chemical Information and Modeling,
2012
167.
Chen L., Gasteiger J.
Journal of the American Chemical Society,
1997
168.
Sello G.
Tetrahedron,
1998
169.
Sello G., Termini M.
Tetrahedron,
1997
170.
Ghiandoni G.M., Bodkin M.J., Chen B., Hristozov D., Wallace J.E., Webster J., Gillet V.J.
Journal of Chemical Information and Modeling,
2019
171.
10.1016/j.mencom.2021.11.003_b0865
Vovk
Algorithmic Learning in a Random World,
2005
172.
10.1016/j.mencom.2021.11.003_b0870
Wei
Sci.,
2016
173.
Struebing H., Ganase Z., Karamertzanis P.G., Siougkrou E., Haycock P., Piccione P.M., Armstrong A., Galindo A., Adjiman C.S.
Nature Chemistry,
2013
174.
Walker E., Kammeraad J., Goetz J., Robo M.T., Tewari A., Zimmerman P.M.
Journal of Chemical Information and Modeling,
2019
175.
C. Coley, M. Fortunato, H. Gao, P. Plehiers, M. Cameron, M. Liu, Y. Wang, T. Struble, J. Liu and Y. Mo, GitHub, 2021, https://github.com/ASKCOS.
176.
T. N. Kipf and M. Welling, arXiv: 1609.02907, 2016.
177.
Korolev V., Mitrofanov A., Korotcov A., Tkachenko V.
Journal of Chemical Information and Modeling,
2019
178.
Jaeger S., Fulle S., Turk S.
Journal of Chemical Information and Modeling,
2018
179.
Zhavoronkov A., Ivanenkov Y.A., Aliper A., Veselov M.S., Aladinskiy V.A., Aladinskaya A.V., Terentiev V.A., Polykovskiy D.A., Kuznetsov M.D., Asadulaev A., Volkov Y., Zholus A., Shayakhmetov R.R., Zhebrak A., Minaeva L.I., et. al.
Nature Biotechnology,
2019
180.
Sattarov B., Baskin I.I., Horvath D., Marcou G., Bjerrum E.J., Varnek A.
Journal of Chemical Information and Modeling,
2019
181.
Merk D., Friedrich L., Grisoni F., Schneider G.
Molecular Informatics,
2018
182.
10.1016/j.mencom.2021.11.003_b0920
Popova
Sci. Adv.,
2018
183.
Sanchez-Lengeling B., Aspuru-Guzik A.
Science,
2018
184.
Coley C., Jin W., Rogers L., Jamison T.F., Jaakkola T.S., Green W.H., Barzilay R., Jensen K.F.
Chemical Science,
2019
185.
Karpov P., Godin G., Tetko I.V.
Lecture Notes in Computer Science,
2019
186.
187.
Asche S., Cooper G.J., Keenan G., Mathis C., Cronin L.
Nature Communications,
2021
188.
Steiner S., Wolf J., Glatzel S., Andreou A., Granda J.M., Keenan G., Hinkley T., Aragon-Camarasa G., Kitson P.J., Angelone D., Cronin L.
Science,
2019
189.
Henson A.B., Gromski P.S., Cronin L.
ACS Central Science,
2018
190.
Burger B., Maffettone P.M., Gusev V.V., Aitchison C.M., Bai Y., Wang X., Li X., Alston B.M., Li B., Clowes R., Rankin N., Harris B., Sprick R.S., Cooper A.I.
Nature,
2020
191.
Vaucher A.C., Zipoli F., Geluykens J., Nair V.H., Schwaller P., Laino T.
Nature Communications,
2020
192.
Dial-a-Molecule EPSRC Grand Challenge Network Website, 2021, http://generic.wordpress.soton.ac.uk/dial-a-molecule/.